
What is sickle cell anaemia?
Image credit: Keith Chambers / via Wikimedia Commons

Sickle cell anaemia is an inherited blood cell condition in which red blood cells develop atypically.
- Sickle cell anaemia is a recessive genetic condition that causes a person’s haemoglobin – the protein that carries oxygen around the body – to become hard and sticky. This can cause the red blood cells to get stuck in blood vessels, leading to pain and other health problems.
- It’s caused by a mutation in the HBB gene, found on chromosome 11.
What is sickle cell anaemia?
- Sickle cell anaemia is caused by a mutation in a gene called haemoglobin beta (HBB), located on chromosome 11.
- It is a recessive genetic disease, which means that both copies of the gene must contain the mutation for a person to have sickle cell anaemia.
- If an individual has just one copy of the mutated gene, they are said to be a carrier of the sickle cell trait. If both biological parents are carriers, there is a chance their child could be born with sickle cell anaemia.
- We now know that sickle cell trait can also make the individual more resistant to malaria. This is because the mutation affects the haemoglobin enough to make it resistant to infection by the malaria-causing parasite, but not so much that it blocks the flow of blood.
What is the biology of sickle cell anaemia?
- The HBB gene codes for haemoglobin, a protein in red blood cells that carries oxygen around the body.
- A mutation in HBB results in a change in one of the bases in the genetic sequence from an A to a T. This changes the amino acid in the haemoglobin protein from glutamic acid to valine.
- The body then produces a new form of haemoglobin called HbS, which behaves differently to regular haemoglobin (HbA).
- HbS causes the red blood cells to become sickle-shaped (rather than the typical doughnut shape), harder and less flexible. This means that they can become stuck in the blood vessels, causing blockages.
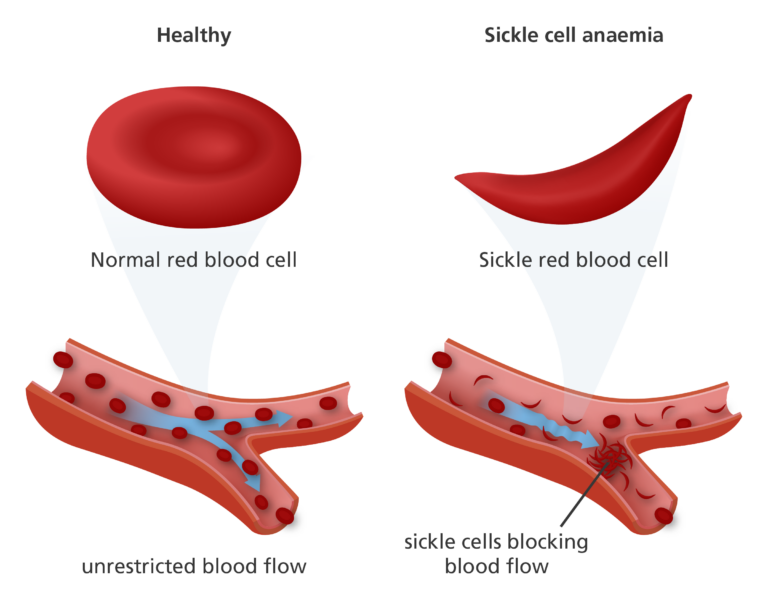
What are the symptoms of sickle cell anaemia?
- The symptoms of sickle cell anaemia vary considerably from person to person.
- Pain can develop when sickle-shaped red blood cells block the flow of blood to the chest, abdomen and joints. These spells of pain are called ‘sickle cell crisis’ and can last anything from a few minutes to several months.
- Symptoms can have a significant impact on quality of life and can lead to life-threatening complications such as:
- stroke: where the supply of blood to the brain becomes blocked.
- acute chest syndrome: where the lungs suddenly lose their ability to breathe in oxygen as a result of sickle cells blocking blood vessels in the lungs.
- increased risk of infection: sickle cell anaemia can damage the spleen, a key organ involved in fighting infection.
- pulmonary hypertension: where sickle-shaped red blood cells block the flow of blood from the heart to the lungs causing the blood pressure in these vessels to become dangerously high.
- Sudden deterioration may be characterised by a high body temperature, severe, uncontrollable pain or difficulty breathing.
- Methods to deal with sickle cell anaemia have improved dramatically in recent years – today, serious complications rarely occur, and people now live much longer than they used to. Just 40 years ago, many people died before the age of two by bacterial infection.
How is sickle cell anaemia diagnosed?
- Sickle cell anaemia is diagnosed using a blood test which detects the presence of the abnormal HbS haemoglobin in the red blood cells.
- In children the blood is taken by pricking a finger or heel. In adults the blood is drawn from a vein in the arm.
How is sickle cell anaemia treated?
- Most treatments aim to treat the individual symptoms. Treatment plans require a number of different healthcare professionals working together, such as haematologists (specialists in blood disorders), clinical psychologists, social workers and physiotherapists who can help patients with pain monitoring and relief.
- For example, regular blood transfusions can help reduce the risk of complications like stroke, by up to 90%. Chelation therapy may need to be given after transfusion to remove excess iron from the patient’s body, which can lead to complications including liver cancer, diabetes and infections.
- Daily antibiotics, such as penicillin, are often given to help protect against serious infections in children aged under five years.
- Pain relief is provided to reduce the pain associated with sickle cell crisis. Lifestyle advice, such as drinking plenty of fluids, can also help reduce the risk of sickle cell crisis.
- If individuals continue to experience pain, a daily medication called hydroxycarbamide may be offered, which stimulates the body to produce another type of haemoglobin, called foetal haemoglobin. Foetal haemoglobin is not affected by the mutation that causes sickle cell anaemia and can carry oxygen around the body and help reduce the risk of a sickle cell crisis occurring.
- The risk of an individual developing complications can also be assessed to help prevent them occurring. For example, a brain scan can measure the rate of blood flow in blood vessels in the head and neck, which supply blood to the brain. Narrow arteries can increase the risk of the person having a stroke in the future.
- Blood and bone marrow transplants may offer a cure for a small number of people.
Is it possible to screen for sickle cell anaemia?
- In the UK, all pregnant people are asked to answer a questionnaire to assess their risk of carrying a baby with sickle cell anaemia. Both biological parents may then be offered a blood test to find out if they are a carrier of the HBB mutation that causes sickle cell anaemia. If both parents are carriers, there is a chance the baby will be born with sickle cell anaemia.
- All babies in the UK are tested for sickle cell anaemia after birth through the heel-prick newborn screening test.
- Anyone from at risk groups having a general anaesthetic is tested for sickle cell anaemia. This is because general anaesthetic reduces the amount of oxygen in the blood, which could be dangerous for someone with the condition.